


2025-03-31 14:55:24
First, let's talk about what HGVS is.
HGVS stands for Human Genome Variation Society. This organization is an affiliate of the International Federation of Human Genetics Societies and the Human Genome Organization (HUGO). Its primary role is to discover and classify human genome variations, including those associated with population distribution and phenotypes, and to update data and related clinical variations in line with advancements in methodology and informatics. The variant naming rules established by this society are the most widely used.
The naming of sequence variants must adhere to the principles of precision, clarity, and stability, with sufficient flexibility to describe all known types of variants. In 2000, HGVS proposed a comprehensive set of variant naming guidelines, which were widely accepted in the academic community and became the standard naming rules in the field of molecular diagnostics. In 2015, HGVS released a new version, HGVS15.11, which corrected errors in the previous version, removed cumbersome expressions, and added naming rules for complex mutations.

Let's delve into the details of these guidelines.
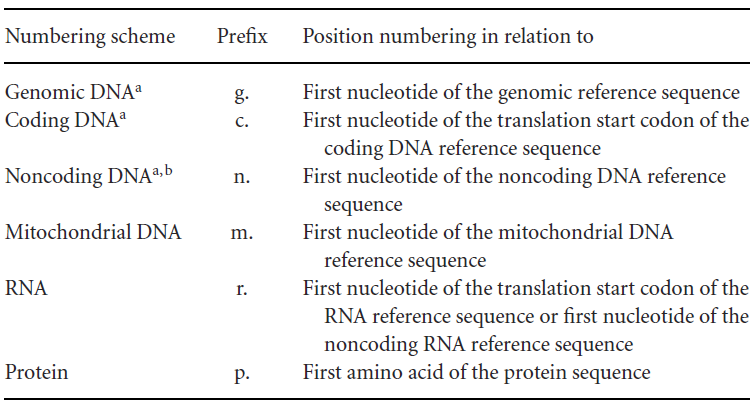
First, reference sequences are divided into different levels, each with its own notation form, distinguished by prefix letters, such as:
g. for genomic reference sequence
c. for coding DNA reference sequence (starting from the start codon)
n. for non-coding DNA reference sequence
m. for mitochondrial DNA reference sequence
r. for RNA reference sequence
p. for protein reference sequence
Each level of sequence has different types of variants, which are listed below:
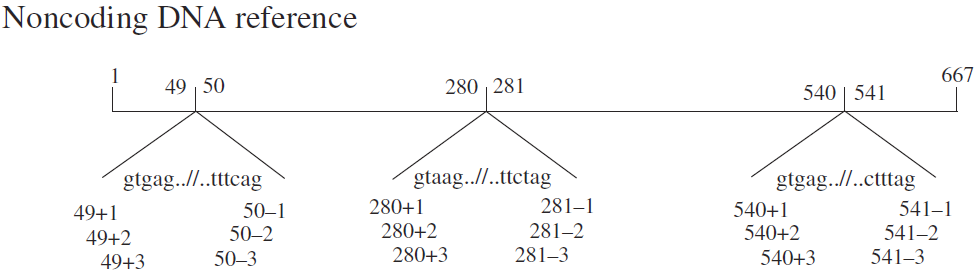
Substitution (>): One nucleotide is replaced by another nucleotide.
For example, g.1318G>T indicates that at the genomic level, the G at position 1318 is replaced by T.
Deletion (del): One or more nucleotides are missing.
For example, g.3661_3706del indicates that at the genomic level, the sequence from position 3661 to 3706 is deleted.
Inversion (inv):
Multiple nucleotides are replaced by their reverse complementary nucleotides (essentially, a chromosomal inversion has occurred).
For example, g.495_499inv indicates that at the genomic level, the sequence from position 495 to 499 has been mutated to its reverse complementary sequence.

Duplication (dup): One or more identical nucleotides are inserted at the 3' end of the variant position (essentially, the reference sequence has been duplicated).
For example, g.3661_3706dup indicates that at the genomic level, the sequence from position 3661 to 3706 has been duplicated.
Insertion (ins): One or more nucleotides are added to a specific position, and these nucleotides are different from the sequence at that position (to distinguish from dup).
For example, g.7339_7340insTAGG indicates that at the genomic level, a TAGG sequence has been inserted between positions 7339 and 7340.
Translocation (con): A segment of nucleotides at the variant position is replaced by a segment from another region of the genome.
For example, g.333_590con1844_2101 indicates that at the genomic level, the sequence from position 333 to 590 has been replaced by the sequence from position 1844 to 2101.
Deletion/Insertion (delins/indel): One or more nucleotides are replaced by one or more other nucleotides, and this does not fall under substitution, inversion, or translocation.
For example, g.112_117delinsTG indicates that at the genomic level, the sequence from position 112 to 117 has been deleted and replaced by a TG sequence.
For the numbering of sequences, HGVS also has strict notation rules, and different levels of variation have different numbering methods:
①Genomic reference sequence: Numbering starts from the first nucleotide of the reference sequence, starting from "1," indicated as g.1, g.2, g.3, without using symbols such as "+," "-," or "*."
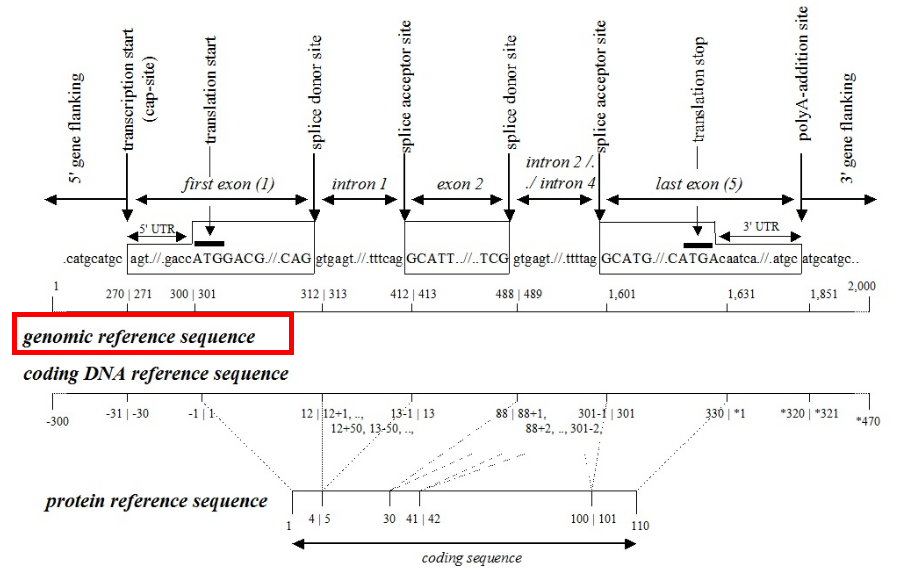
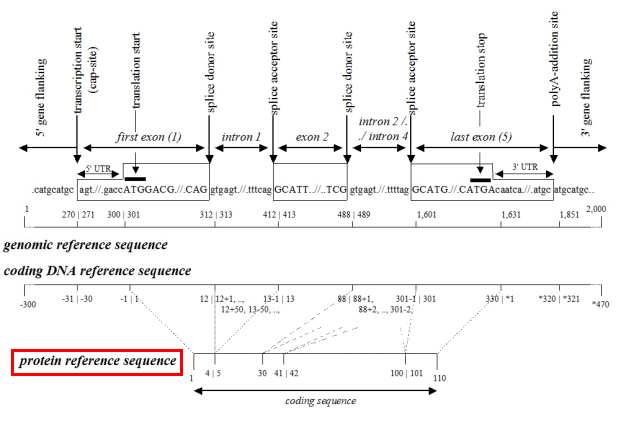
②Coding sequence: In the field of molecular diagnostics, using the coding sequence to represent variants is more common because this method provides specific positional information (exon/intron, start codon/stop codon, amino acid number of the variant).
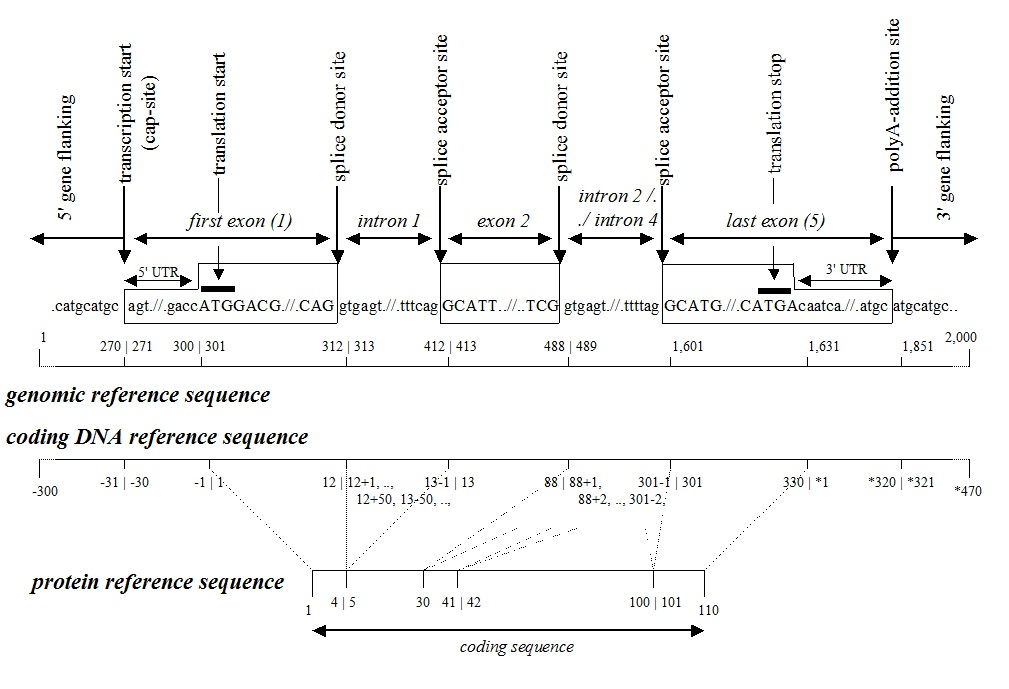
A.Coding sequence level: Numbering starts from the start codon, beginning with "1," and exon numbers are continuous. Introns and UTRs are not numbered. The region upstream of the start codon is indicated as c.-1, c.-2, c.-3, etc., and the region downstream of the stop codon is indicated as c.*1, c.*2, c.*3, etc.
B.Intron region: Variants in the intron are numbered based on the nearest exon. For variants near the 5' end of the intron, the position is numbered based on the upstream exon. For example, c.187+1 indicates that the last nucleotide of the upstream exon is 187, and the variant is the first nucleotide at the 5' end of the intron. For variants near the 3' end of the intron, the position is numbered based on the downstream exon. For example.
c.188-1 indicates that the first nucleotide of the downstream exon is 188, and the variant is the first nucleotide at the 3' end of the intron.
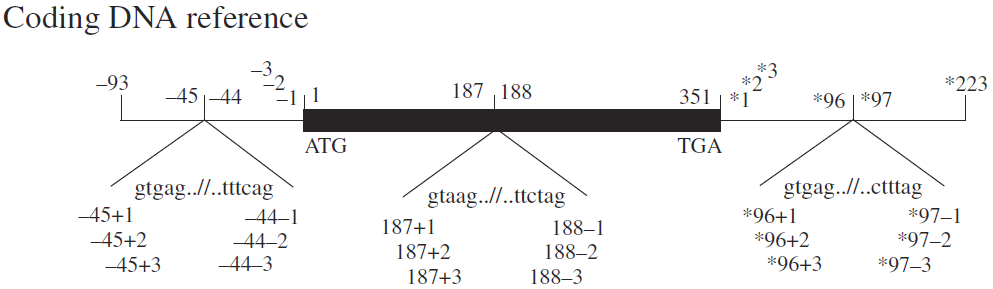
C.UTR numbering: Similar to introns, such as c.-123, c.*345, etc. For introns within the UTR, similar rules apply. For example, c.-55+23 indicates that the last nucleotide of the upstream exon is -55 (the 55th nucleotide upstream of the start codon), and the variant is the 23rd nucleotide at the 5' end of the intron. *c.55-23 indicates that the first nucleotide of the downstream exon is 55 (the 55th nucleotide downstream of the stop codon), and the variant is the 23rd nucleotide at the 3' end of the intron.
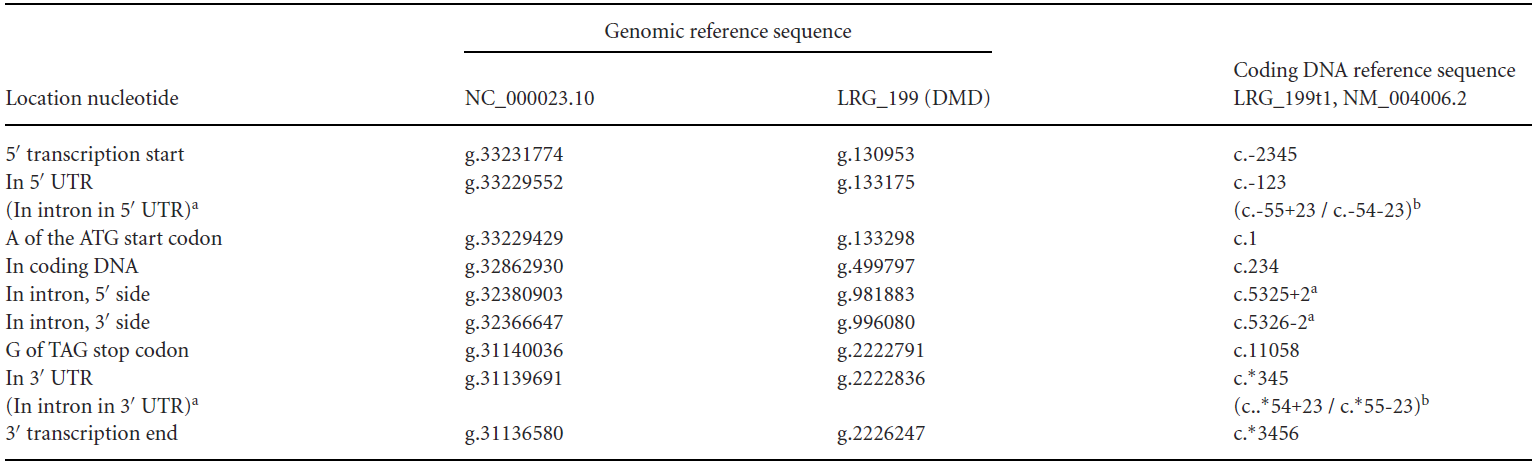
③Non-coding sequence: Numbering of non-coding DNA starts from the first nucleotide of the reference sequence, beginning with "1," indicated as n.1, n.2, n.3. Introns are not numbered, and the notation for intron regions is the same as for coding sequences.
The above are the notation rules for different levels of DNA.。
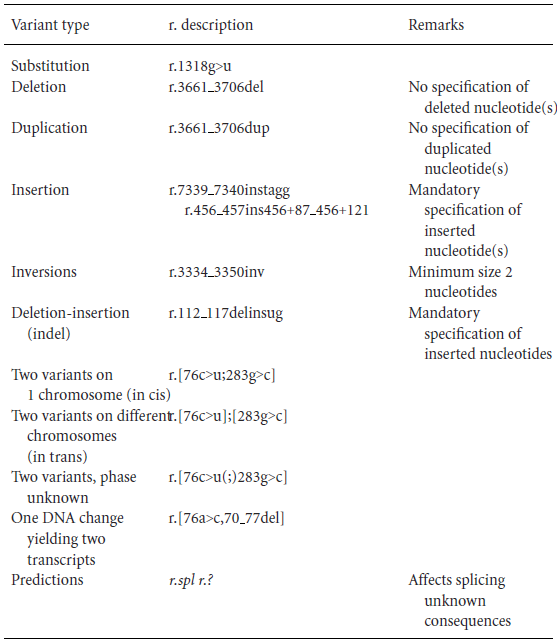
For RNA-level variants, the notation is similar to that of DNA. Once you understand the DNA rules, you can easily understand RNA notation. For example:
r.1318g>u
r.3661_3706del
r.3661_3706dup
r.112_117delinsug
The difference from DNA is that the nucleotide sequence is indicated in lowercase letters, and "u" is used instead of "t."
Specific examples are provided below:
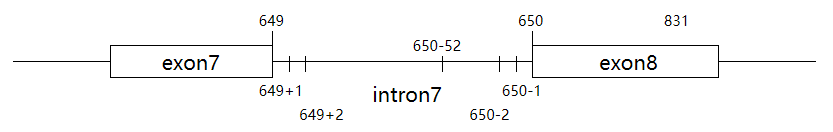
A.RNA-level notation—splice site mutation (splicing variant)For example, a coding sequence variant c.650-1G>C may cause the following splicing changes:①r.649_650ins[650-52_650-2;g>c] indicates that at the RNA level, the nucleotide sequence from position 2 to 52 of the 3' end of intron 7 is inserted between nucleotides 649 and 650 of the transcript RNA. The first nucleotide of the 3' end of intron 7 has undergone a G-to-C mutation. The essence of this variant is that the first nucleotide of the 3' end of intron 7 has mutated, causing a change in the initial transcript RNA splicing site (splicing occurs between nucleotides 52 and 53 of the 3' end of intron 7). A segment of intron 7 (nucleotides 2 to 52 of the 3' end) is retained in the mature transcript.
B.RNA-level notation—splice site mutationFor example, a coding sequence variant c.650-1G>C may cause the following splicing changes:
②r.650_831del indicates that at the RNA level, the nucleotide sequence from position 650 to 831 of exon 8 is deleted. The essence of this variant is the same as in example A, where the first nucleotide of the 3' end of intron 7 has mutated, causing a change in the initial transcript RNA splicing site (splicing occurs between nucleotides 831 and 832 of exon 8). As a result, a segment of exon 8 (nucleotides 650 to 831) is missing in the final mature transcript RNA.
Finally, let's look at the most common and easiest-to-understand protein-level changes:
Protein reference sequence: Numbering starts from the first amino acid, beginning with "1." Variant amino acids are indicated using standard abbreviations (either three-letter or one-letter codes).For the most common and easily understandable protein-level variant notations, there are several examples:
①Amino acid mutation:p.Arg490Ser or p.R490S indicates that at the protein level, the arginine at position 490 has mutated to serine.
BRAF V600E indicates that the valine at position 600 of the BRAF protein has mutated to glutamic acid.
EGFR T790M indicates that the threonine at position 790 of the EGFR protein has mutated to methionine.
②Nonsense mutation (*):
p.Trp78Ter or p.Trp78* indicates that at the protein level, the tryptophan at position 78 has mutated to a stop codon (nonsense mutation).
Frameshift mutation (fs):
p.Arg97ProfsTer23 indicates that at the protein level, a frameshift mutation has occurred. The arginine at position 97 has mutated to proline, and renumbering starts from position 97 (starting from 1). The 23rd amino acid after the frameshift mutation has become a stop codon.
③Insertion/deletion:
p.Asp388_Gln393del indicates that at the protein level, five amino acids from position 388 (aspartic acid) to position 393 (glutamine) have been deleted.

In addition to the above, a complete HGVS variant name must also include the variant reference sequence. The reference sequence uses IDs from public databases such as NCBI (National Center for Biotechnology Information) or EBI (European Bioinformatics Institute), including the accession number and version number of the sequence, such as NC_# (e.g., NC_000023.10), NG_# (e.g., NG_012232.1), NM_# (e.g., NM_004006.2), etc.If a variant cannot be found in the above reference genomes, the Locus Reference Genomic sequence (LRG) database should be selected, such as LRG_199, LRG_304, etc.
The SVD-WG is considering using reference sequences from the Ensembl database, such as ENSG00000182533.6, ENST00000357033.8, ENSP00000354923.3, etc. One main issue is that the reference sequences downloaded from the Ensembl database do not include version numbers.
A complete variant notation example is:NG_012232.1(NM_004006.2):c.357+1G>ANC_000023.10(NM_004006.2):c.357+1G>ALRG_199t1:c.357+1G>ANCBI RefSeq Accession NumbersNCBI's RefSeq (Reference Sequence) is a free database that collects DNA, RNA, and protein sequences. RefSeq provides a large amount of genomic, transcript, and translation product sequence information for multiple species.Each given sequence in RefSeq has a unique accession number, which is usually written in the form of NN_NNNNN.N. The prefix NN is used to distinguish the source of the sequence (genomic sequence, transcript sequence, or protein sequence, etc.), the middle NNNNN is a number of varying length that encodes the sequence, and the final N is the version number of the sequence. The original version number is 1, and new versions increment the original number by 1 to indicate that the sequence information has been updated compared to the previous version.
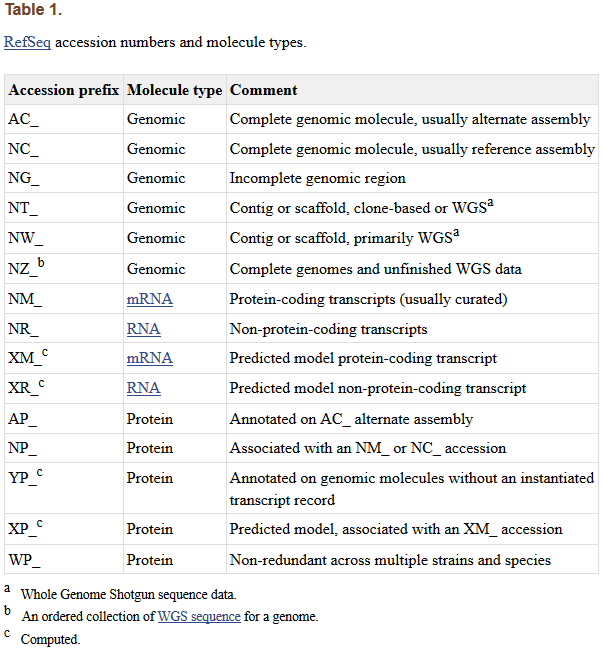
Common classifications of accession number prefixes include:AC: Complete, annotated genomic sequences (mainly for viruses or prokaryotes).
NG: Incomplete genomic regions (non-transcribed pseudogenes or difficult-to-annotate genomic regions).
NC: Complete, reference genomic sequences.
NM: Coding transcripts (usually verified sequences).
NR: Non-coding transcripts.
NP: Protein sequences, corresponding to NM or NC.

For those unable to find a reference genome in NCBI, the Locus Reference Genomic (LRG) database can be referenced. In 2008, EMBL-EBI, NCBI, HGVS, and other groups established the LRG plan based on the RefSeqGene initiative, aiming to establish a universal reference standard for variant reporting. The new plan addresses the shortcomings of the existing variant notation system (including the consistency of reference sequence version numbers) and provides a clear, explicit, and consistent set of rules for reporting clinically relevant gene locus variants.LRG integrates Ensembl, NCBI, and UCSC genome browsers to provide genome visualization and analysis, covering existing annotation information from each database. The genome versions include GRCh37 and GRCh38. LRG uses a more streamlined reference sequence numbering system, typically using a single ID to represent a variant gene (avoiding the hassle of changes in reference sequence version numbers). Clinical variant reporting is manually supervised and annotated by experts.
Once you become proficient in these naming rules, you will not only be able to easily understand various mutations in the report but also be able to look up relevant information according to your needs and obtain more useful data. It is hoped that this article will be helpful to everyone!
References:
1.Johan T. den Dunnen et al HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 20162.varnomen.hgvs.org/3.lrg-sequence.org/4.grch37.ensembl.org/5. ncbi.nlm.nih.gov/
